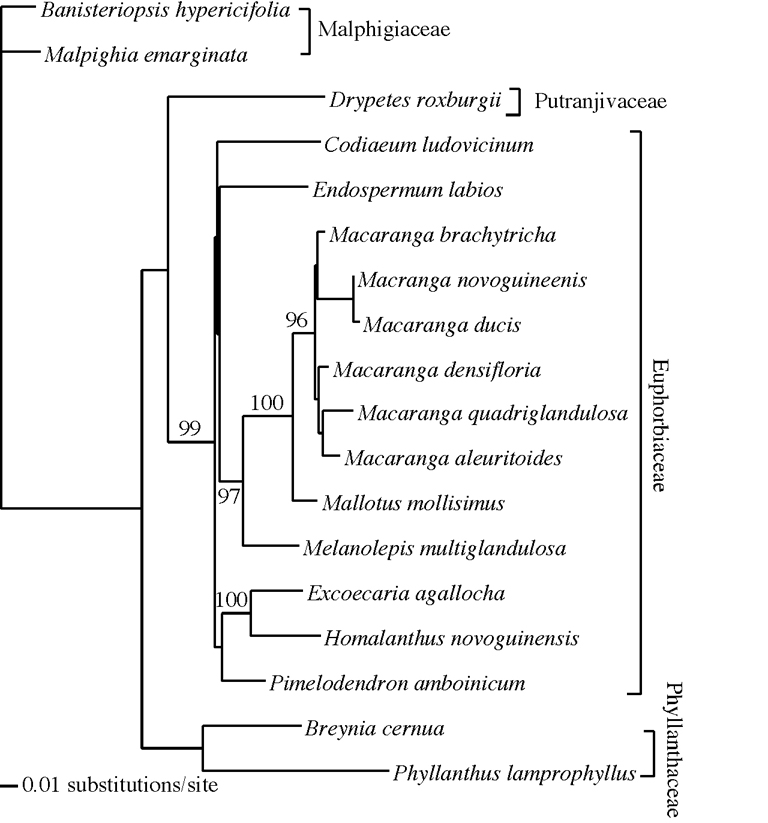
Appendix A. Euphorbiaceae phylogeny.
Euphorbiaceae DNA was extracted from 10–15 mg silica gel preserved leaf fragments using the Qiagen DNeasy Plant Extraction kit (Valencia, California, USA). PCR amplification of 2100 bp was performed in two reactions of ~1200 bp each using primers from Olmstead and Sweere (1994) including ndhF8f, ndhF 972, ndhF1318r, and ndhF2110r. Amplification conditions were ~20 ng genomic DNA, 5 uL Qiagen PCR buffer, 160 nM each primer, 0.2 mM each dNTP, and QIAGEN Hotstart Taq. Thermal cycling included a 15 min hold at 95°C followed by 25 cycles of 94°C for 30 s, 48°C for 60 s, 68°C for 90 s, and a final extension at 72°C for 7 min. PCR products were cleaned using the QIAquick or MinElute PCR purification spin columns (Qiagen, Valencia, California, USA). Cleaned PCR products were quantified using Hoechst 33258 fluorescent dye in a Turner Quantech fluorometer. Ten µL sequencing reactions were performed with Big Dye sequencing reagents and protocols (versions 2 and 3; Applied Biosystems; Foster City, California, USA) and data were collected using an ABI 377 Automated DNA sequencer. Between seven and nine sequencing primers were used for each taxon, including the aforementioned primers, and ndhF972r, ndhF1318, and ndhF1603r. Sequences were edited using Sequencher v. 3.0 (Gene Codes Corp., Ann Arbor, Michigan, USA). Sequence alignment was performed manually to maintain open reading frames for the entire portion of the gene.
Tree searches were performed using PAUP* 4.0b10 (Swofford 2002) with the TBR branch-swapping algorithm and 10,000 random addition sequence replicates. Maxtrees was set to increase without limit. Support was assessed with 1,000 bootstrap replicates (10 addition sequence replicates per bootstrap replicate), and maxtrees set at 10,000. Clade support and tree length calculations were conducted with uninformative characters excluded. Banisteriopsis hypericifolia (Malphigiaceae), Malpighia emarginata (Malphigiaceae), and Drypetes roxburgii (Putranjivaceae) served as outgroups for rooting purposes (Davis et al. 2001) .
Modeltest version 3.4 (Posada and Crandall 1998) was used to select an appropriate substitution model for maximum likelihood analyses. Likelihood searches were performed using PAUP* 4.0b10 (Swofford 2002) with a neighbor joining tree as a starting topology and model parameters obtained from Modeltest. Parameters were re-estimated using the tree with the highest likelihood. These new parameter estimates were used in an additional round of TBR branch swapping on the maximum likelihood (ML) tree to obtain a tree with higher likelihood. Branch swapping and parameter estimation were iterated in this fashion until subsequent analyses converged on the same likelihood score and model parameters.
Euphorbiaceae ndhF sequences (Genbank accession numbers AY374311- AY374325) provided phylogenetic resolution within the family. Out of 1,160 aligned positions, 728 (63%) were constant, 230 (20%) were apomorphic, and 202 (17%) were parsimony-informative. Heuristic searches constrained by the monophyly of outgroup Malphigiaceae yielded 360 equally parsimonious trees of 433 steps (CI = 0.674; RI = 0.733 excluding uninformative sites). Nonparametric boostrapping strongly supported the monophyly of Macaranga , close relationships of Mallotus and Melanolepis to Macaranga , the sister relationship of Excoecaria and Homalanthus , and the monophyly of the Euphorbiaceae excluding Phyllanthaceae (Fig. A1). A heuristic search under maximum likelihood yielded two topologies of equal likelihood that were identical to two of the most parsimonious trees. The GTR+ G rate matrix for the topology shown in Fig. A1 was AC = 1.07271, AG = 1.83838, AT = 0.35044, CG = 1.54686, CT = 2.34400 and a = 0.664333. Estimated base frequencies were A = 0.31201, C = 0.14941, and G = 0.158714.
|
| FIG. A1. A maximum-likelihood phylogram of New Guinea Euphorbiaceae and Phyllanthaceae (-lnL = 4889.09 with GTR+ G model parameters estimated from the topology shown). The tree was rooted with Malphigiaceae. Nodes with > 50% non-parametric bootstrap percentages under parsimony are labeled. |
LITERATURE CITED
Davis, C. C., W. R. Anderson, and M. J. Donoghue. 2001. Phylogeny of Malpighiaceae: evidence from chloroplast ndhF and trnl-F nucleotide sequences. American Journal of Botany 88:1830–1846.
Olmstead, R. G., and J. A. Sweere. 1994. Combining data in phylogenetic systematics: an empirical approach using three molecular data sets in the Solanaceae. Systematic Biology 43:467–481.
Posada, D., and K. A. Crandall. 1998. Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818.
Swofford, D. L. 2002. PAUP*: phylogenetic analysis using parsimony (*and other methods), version 4. Sinauer Associates, Sunderland, Massachusetts, USA.